

 联系我们
联系我们
药物靶点的重要性不言而喻,其价值不仅体现在能够发表证明特定药物靶点存在的科研文章,更重要的是,明确药物靶点后,可以针对这一靶点设计新型药物。对于已知药物,我们可以在确保其与蛋白质结合靶点不变的前提下,对其非结合部位进行化学修饰,从而在保持药物疗效的同时减少或避免副作用,这正是药物靶点筛选的核心价值所在。
一、药物靶点证明关键点
如何证明药物靶点?这一过程与蛋白分子互作的研究既有相似之处,也存在差异。主要可以从以下两个方面进行阐述:
1.证明分子间的相互作用
蛋白分子互作:主要集中在细胞内,研究涉及信号通路相关蛋白之间的相互作用,尤其是蛋白与蛋白之间的相互作用。
药物靶点:不仅涉及蛋白与蛋白之间的相互作用(如单克隆抗体),还包括多肽与蛋白的相互作用,以及小分子药物(如中药单体成分或其他抑制剂)与蛋白的结合。因此,证明药物靶点的方法更为多样和复杂。
2.结合强度的论证
蛋白分子互作:只要证明分子之间有结合即可,结合强度的论证相对简单。通常通过免疫沉淀(IP)、pull down 和突变互作位点等体外实验来证明分子间的结合。比如分子相互结合后去激活下游通路,这种不需要有很强的结合力。
药物靶点:药物与靶蛋白的结合强度,即解离常数(KD),是个关键因素。一个药物即使能够与靶蛋白结合,但如果结合不稳定,其效果可能非常有限。因此,药物的结合强度需要通过多种方法进行严格验证,确保其在生理条件下能够稳定发挥作用。
二、药物靶点筛选和证明的实验思路
1.筛选
1.1,FRET(荧光共振能量转移,药物筛选)
1.2,ABPP(基于活性蛋白质组学分析,靶点筛选)
1.3,Lip/MA(限制性酶解-质谱分析,靶点筛选)
2.证明结合
2.1,CETSA(细胞热位移分析;温度)
2.2,DARTS(药物亲和反应靶向稳定性技术;酶)
2.3,荧光共定位
2.4,Pull down
2.5,SPR(表面等离子共振)/MST(微尺度热涌动)
2.6,分子对接(计算机模拟靶点)
2.7,突变体构建(具体靶点)
3.功能验证
3.1,Gene KO/KD/OE
3.2,通路验证/细胞表型/疾病表型验证
1.1 筛选药物
在筛选过程中,荧光共振能量转移(FRET)方法用于已知影响疾病或细胞表型的关键通路,但未知哪些药物能作用于该通路时,筛选潜在药物。通过 FRET 技术,可以发现能够影响关键蛋白活性的药物,然而,这种方法并不能直接证明药物的靶点即为该关键蛋白。
1.2-1.3筛选靶蛋白
最常用且有效的药物靶点确认方法包括:基于活性蛋白质组学分析的ABPP及通过限制性酶解将蛋白分解为多肽片段后进行质谱分析的Lip/MA。通过蛋白质组学定量分析可确定药物富集最多的靶点,并逐步验证。
2.1-2.5证明有结合
细胞热位移分析(CETSA)和药物亲和反应捕获稳定化实验(DARTS)均可通过药物与靶蛋白的结合,分别减轻靶蛋白的高温降解和蛋白酶降解程度。这两种方法还可与质谱联用,用于药物靶点的筛选。
此外,荧光共定位可证实药物与靶蛋白在空间上的结合,而Pull down、表面等离子体共振(SPR)、微尺度热泳动(MST)等技术则用于验证二者是否直接结合。其中,Pull down 实验可以通过纯化蛋白或在体内过表达蛋白后检测它们的结合情况,而 SPR 和 MST 则是在体外条件下证明药物与靶蛋白的直接结合。
2.6-2.7结合具体靶点
分子对接和突变体构建用于验证具体的结合靶点。通过计算机模拟筛选活性位点,活性位点通常包含多个肽段。制备截短突变体,确定靶点所在的肽段,随后对该肽段进行点突变,以精确定位具体的结合位点。确定该靶点后,不仅可以验证药物的效果,还可以基于靶点信息优化和改良药物。
3.1-3.2功能验证
通过基因敲除(Gene KO)、RNA干扰(Gene KD)和基因过表达(Gene OE)的方法调节靶点的表达水平,进而验证药物对相关表型的影响。这些表型不仅包括相关通路,细胞层面的变化,如细胞死亡或代谢变化,还包括整体代谢及疾病模型的表型,以评估药物结合靶点对疾病发展的影响。
三、文献实例
以2022年在影响影子27.7《CELL METABOLIS》上发表的“Discovery of a potent allosteric activator of DGKQ that amelioratesobesity-induced insulin resistance via the sn-1,2-DAG-PKCe signaling axis”为例。
该文章涉及到的疾病模型是肥胖、胰岛素抵抗、或者是脂质代谢异常,用到多种肝细胞模型,涉及到的代谢通路是脂质代谢的关键通路sn-1,2-DGK/PKCɛ,文章中最终筛选到的药物是白术内酯2(AT II),找到的对应靶点是DGKQ靶点。
文章作者思路图(图1)可以看出:右边是作者发现该药物通过影响一系列通路,改善了胰岛素抵抗,降低了脂质的累计,最终改善肥胖。
左边是作者筛选这个药物的过程,首先,作者关注到PKC/DAG通路,通过荧光共振能量转移(FRET)以及液相色谱联合质谱的方法,找到哪些药物主要影响这个通路两个关键蛋白的活性,最终选择了AT II药物,进一步通过药靶筛选的方法,最终找到了关键靶蛋白DGKQ,通过截短突变和点突变构建的方法以及计算机模拟,找到了药物AT II与靶蛋白DGKQ结合的直接靶点。
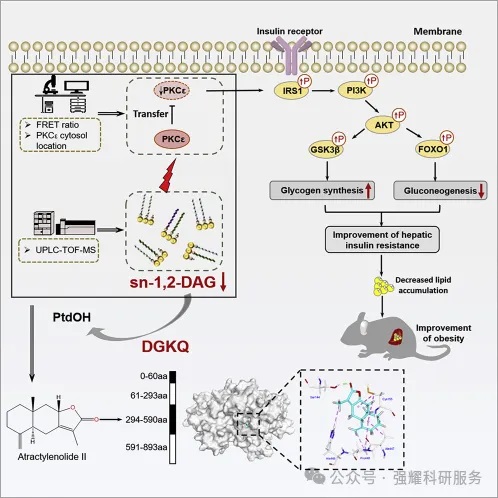
图1 思路图
基于通路/靶点筛选药物的策略具体方法如下
作者在已有了PKC代谢的相关通路,但不知道药物的情况下,通过FRET以及液相色谱联合质谱(图2)的方法,找到影响该通路的关键蛋白就是PKC及DAG,影响这两个蛋白的主要药物最终筛选到AT II,前面筛选到的AT II能影响通路关键蛋白的活性,但并不能证明AT II的靶点就是这蛋白,因此后面需要找关键靶点。

图2 FRET以及液相色谱联合质谱
作者找到最有效的药物后,通过基于活性的蛋白质组学分析(ABPP)(图3)的方法来寻找靶点,它将白术内酯这个小分子化合物结合物素制成一个探针,该探针会与一系列靶蛋白结合,然后通过磁珠将它拉下来,再送蛋白质组学分析,看药物和哪一个片段结合最多,强度最高。
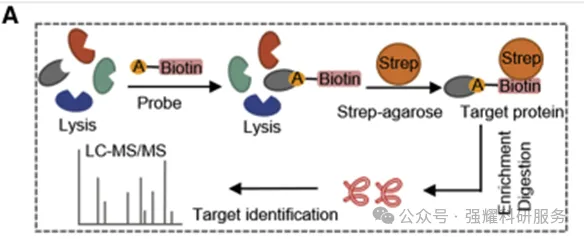
图3 ABPP相关实验
接着是证明药物和靶点是直接结合的。通过pull down(图4) 实验,得到的B、C图表明加入白术内酯II后,可以抑制探针A6与DGKQ的直接结合。接下来MST(图5D)和SPR(图5E)都是证明直接结合,它们实验结果都是微摩尔级,说明它们的结合是非常紧密的。
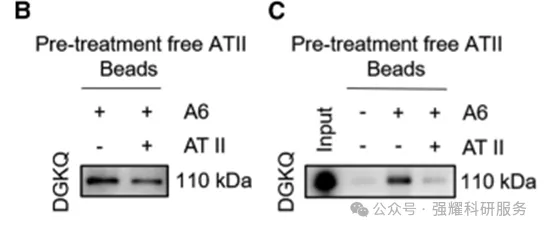
图4 pull down
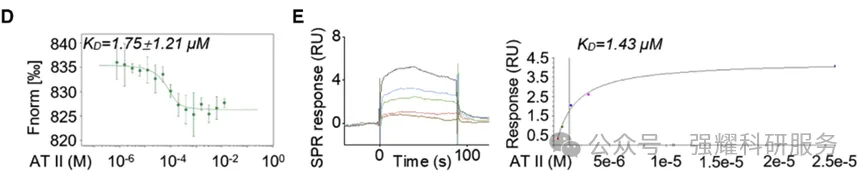
图5 MST和SPR
接下来通过DARTS(图6)和CETSA(图7)技术证明AT II和DGKQ的结合是非常稳定的,它们结合后可以抑制DGKQ被蛋白酶降解,也可抑制DGKQ在高温下降解。
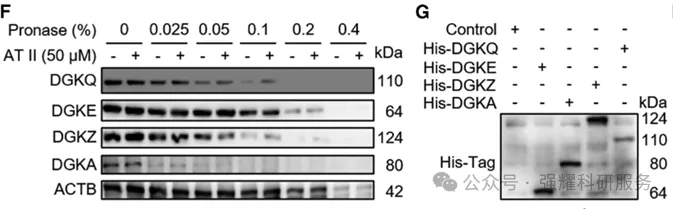
图6 DARTS方法
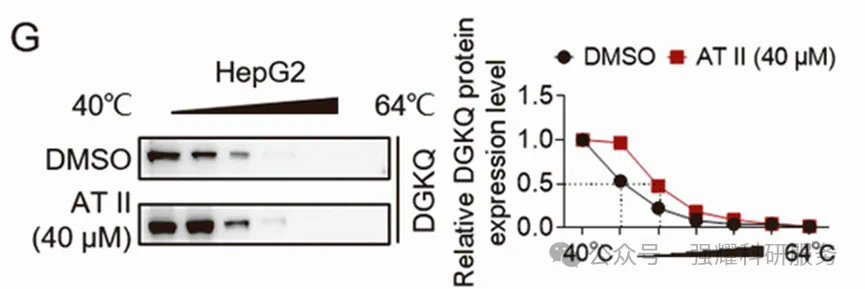
图7 CETSA方法
然后,分析的是AT II的异构体AT I、ATIII、8β-Me,也是通过采用MST和SPR的方法(图8),发现ATII与DGKQ结合最为紧密,其余要么不结合,要么解离常数在10微摩尔级,反向证明了DGKQ只与ATII结合。
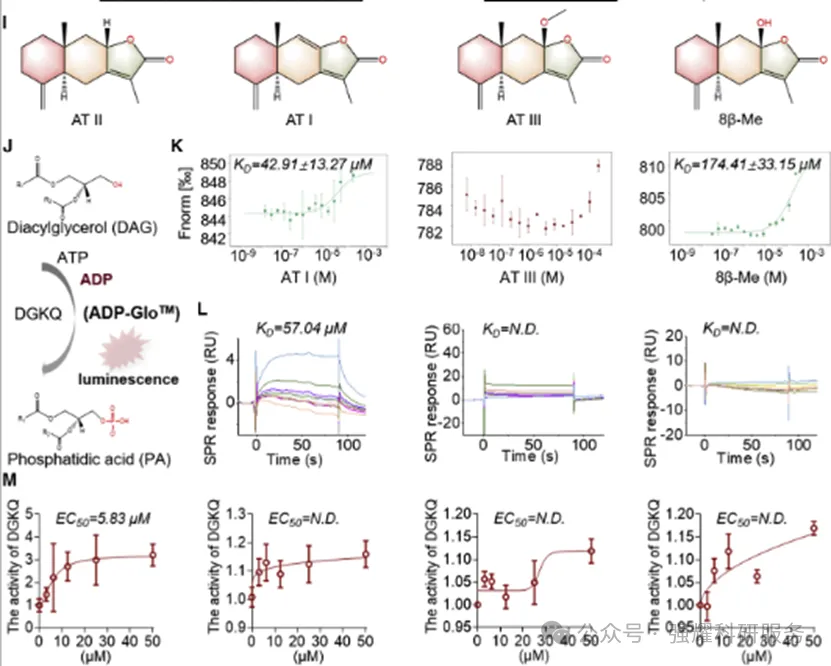
图8 异构体MST和SPR检测
接者是反向验证,或者说是结合靶点验证。首先DGKQ活性位点有很多片段,作者取了其中不同片段来与A6探针结合,Pull down实验结果(图9A)下面图是片段本身大小的图,上面是用A6去拉后得到的这个片段的大小,可以看出61-293aa和294-590aa是与A6有结合。通过MST(图9B),61-293aa的C1和294-590aa的PH片段的解离常数在10微摩尔级,其他片段在100微摩尔级或为0,因此将结合域聚焦在61-590aa区段。
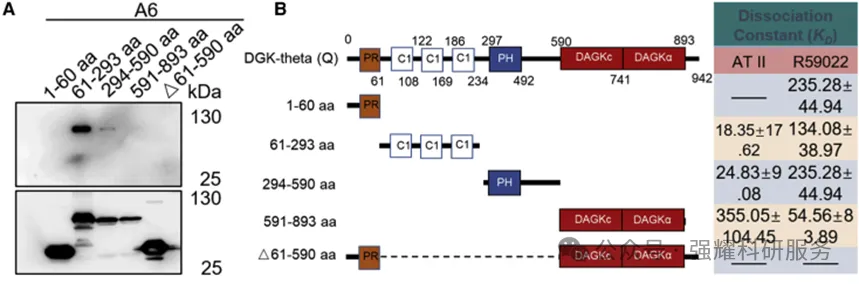
图9 截短突变体Pull down和MST检测
进一步通过分子对接(图10),找到活性位点片段大概结合的一些氨基酸序列,后面构建点突变模型,依旧利用pull down(图11D) 和MST(图11E)确定结合位点在CRD domain 的193,204,241三个氨基酸上。
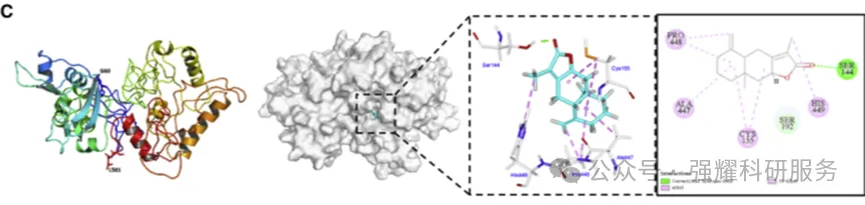
图10 计算机模拟分子对接
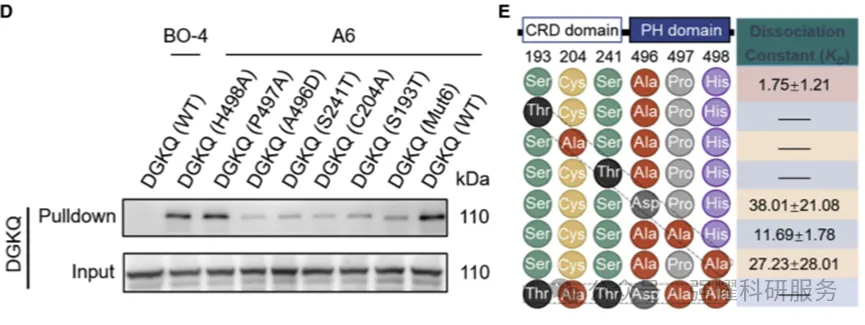
图11 点突变Pull down和MST检测
最后是功能验证,采用了高脂血症模型、肝脏脂质代谢异常模型、胰岛素抵抗模型的小鼠进行验证。
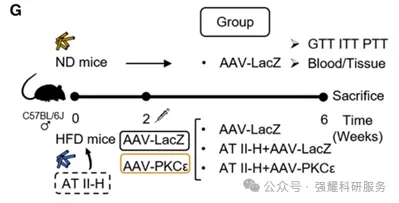
图12 疾病模型功能验证
强耀生物可提供分子互作平台:SPR检测,MST检测,Co-IP,GST-Pull down等服务,欢迎您咨询订购。
 返回
返回